
Miraculous Medicine for Creating Miracles - Structure Activity Analysis of GLP-1/GIP Double Receptor Agonist Tilposide (II)
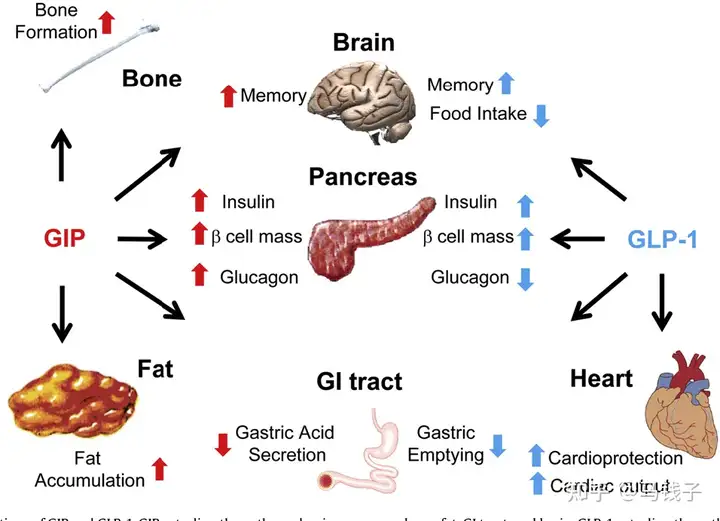
GIP is the first discovered enterotropin, which is currently known as GIP and GLP-1. In the normal human body, the insulin secretion stimulated by the pancreatic stimulating effect (after meals) accounts for approximately 70% of the total insulin secretion, while GIP accounts for two-thirds of the total pancreatic stimulating effect, much higher than GLP-1.
GIP is a single chain peptide hormone composed of 42 amino acid residues, a peptide molecule secreted by K cells in the duodenum and jejunum of the small intestine. The main regulatory mechanism is that after we ingest, nutrients stimulate the small intestine, and endocrine cells (K cells) in the small intestine secrete GIP. After GIP secretion is released into the bloodstream, GIP rapidly increases and reaches its peak within a few minutes. However, its degradation rate is also fast, and the most important thing is that it will be degraded by the ubiquitous degradation enzyme DPP-4 in our body. DPP-4 degrades GIP, just like GLP-1, by cutting off two amino acids at the N-terminus from the peptide chain between the second and third positions of the N-terminus, resulting in a type of degradation product that is inactive/does not have the ability to stimulate insulin secretion. In normal people, the half-life of GIP is about 7 minutes, and even shorter in type 2 diabetes (T2DM) patients. Because the level of DPP-4 enzyme in T2DM patients increases, the activity also increases, so the half-life of GIP is shortened to 5 minutes. After being degraded by DPP-4 enzyme, GIP becomes a 3-42 fragment, without the effect of enterotropin and no longer stimulates insulin secretion. Although it does not have the biological activity to stimulate insulin secretion, it retains its binding effect to the GIP receptor (GIPR), making it equivalent to an antagonist of endogenous GIPR.
The GLP-1 receptor is mainly expressed in β Cells, α The cell does not express GIPR, which is different from it α and β Cells are expressed. Some basic studies have confirmed that the secretion of pancreatic stimulating hormone (GIP) after intestinal intake of nutrients includes GIP. GIP can transmit this signal of nutrient intake to us separately β Cells α Cells. So GIP can directly regulate the bolism of the body [19]. And GIP for α Cell heel β The proportion of cellular regulation is related to the composition of the nutrients we consume: if the nutrients we consume are mainly carbohydrates, GIP will have a direct effect, that is β Perceived by cells. Under the synergistic effect of elevated blood glucose concentration and GIP (GIP acting on GIPR), the β The level of intracellular cyclic adenosine monophosphate (cAMP) can be promoted in a glucose dependent manner β The insulin secretion of cells. In addition, GIP can also upregulate insulin biosynthesis and promote β Cell survival. This includes promoting β Cell proliferation.
GIP is different from GLP-1, except for its β In addition to cellular regulation α Cells also have a direct regulatory effect. And GLP-1 is beneficial for α The regulation of cells is believed by most scholars to be indirect, and GLP-1 may be regulated through β Insulin secreted by cells, or through δ Indirect regulation of cell secretion of somatostatin α The function of cells. Unlike GIP, it can directly α Cells undergo biological regulation because α Cells have clear of GIPR.
GIP actually secretes glucagon α In cells, GIPR is clearly expressed, unlike GLP-1. The regulation of glucagon by GIP is highly refined, depending on different health states of the body and blood sugar levels
Research has shown that GIP can regulate glucagon secretion in a glucose dependent manner in healthy subjects. Specifically, when blood sugar is particularly high, GIP no longer significantly stimulates the secretion of glucagon, while during hyperglycemia, GIP affects β Cells can promote insulin secretion and C-peptide release. On the contrary, GIP can promote fasting or hypoglycemic conditions α The secretion of glucagon by cells.
Although GIP and GLP-1 are both secreted after meals in normal individuals, there is a mutual coordination and interaction between the two,. The combination of GIP and GLP-1 may maximize the effect of enterotropin.
Unfortunately, the producibility of GIP is very low, and in T2DM patients, infusions reaching hyperphysiological GIP concentrations did not trigger significant insulin secretion responses; Therefore, GIP infusion cannot quickly normalize blood sugar levels in T2DM patients. This sluggish response may be caused by the downregulation of GIP receptors (GIPR) by high levels of circulating glucose. However, a large amount of data indicates that drugs that reduce circulating glucose levels can largely overcome GIP resistance, paving the way for considering GIP as a supplement to hypoglycemic therapies such as GLP-1.6. In addition, GIPR signaling blocks vomiting and alleviates other negative side effects of GLP-1 receptor (GLP-1R) activation.
Therefore, d on the above research foundation, multiple pharmaceutical companies are currently developing and researching GIP/GLP-1 dual receptor agonists, such as tilposide.
Tirzepatide is a single molecule bifunctional peptide developed and successfully marketed by Lilly, which can activate both GLP-1R and GIPR simultaneously. Tilposide is composed of 39 amino acids, which are acylated at the C-terminal and bind to the C20 fatty acid moiety through the spacer region of Lys20. Its half-life is 116.7 hours. The affinity between Tilposide and GLP-1R is higher than GLP-1, and its cAMP activation activity is significantly lower. The affinity between tilposide and GIPR is significantly lower than GIP, and its cAMP activity is roughly equal to that of GIP.
In order to achieve dual activity, Tilposide not only fused amino acid residues mainly from GLP-1 and GIP, but also used some unique amino acid residues. In the following text, d on existing reports, analyze how tilposide is constructed.
Through the analysis of the amino acid structure of Tilposide, it was found that its peptide construction mainly comes from GLP-1, GIP, exenatide, and smeglutide, with a few residues being unique.
Tilposide and GLP-1 receptor
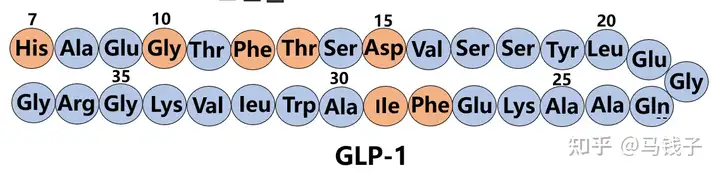

Early structure-activity studies of endogenous GLP-1 have revealed key sequence domains related to potency and enzymatic degradation. Studies have shown that N-terminal residues His7, Gly10, Phe12, Thr13, and Asp15 are crucial for interactions with GLP-1 receptors. Phe28 and Ile29 may be more important for the secondary structure of peptides and therefore for the conformation of receptor recognition, rather than participating in receptor interactions. Therefore, during the construction of Tilposide, Gly4 (Gly10), Phe6 (Phe12), Thr7 (Thr13), Asp9 (Asp15), Phe22 (Phe28) were retained to ensure GLP-1 receptor activity.
Tilposide and GIP receptor activity
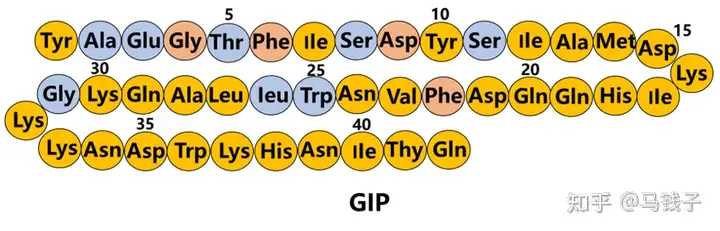
Tyr1:
Tyr1 in GIP may sacrifice its GLP-1 activity while supporting its GIP activity.
Aib2:
GLP-1 (7-37) Ala8 and GIP-Ala2 are DPP-4 cleavage sites, so many studies have focused on reducing this degradation by replacing or modifying the first three residues., The use of Aib2 may only have the smallest impact on GLP-1 and GIP activity. Although Gly2 affects GLP-1 activity to some extent, overall, exenatide shows better activity than natural GLP-1. Therefore, if the biosynthesis of double agonists, Gly2 may be feasible. In addition, since the instability of Ala2 is caused by the degradation of DPP-4, Ala2 may not need to be altered, which creates steric hindrance to prevent enzyme approach.
Thr7:
GLP-1 and GIP are partially homologous, and binding Thr7 from GLP-1 to Tisepatide, replacing IIe7 in GIP, preserves GLP-1 activity and may slightly reduce its GIP activity.
Tyr10、Ile12、Asp 15、Lys16、IIe17、Gln19、Val23、Ala28:
Tilposide retains the highly active amino acid residues of GIP. The positions 10, 12, 13, and 14 of exenatide (a full GLP-1R agonist) are different from those of GLP-1, indicating that these residues are not important for GLP-1. Mutations at positions 16, 17, 18, and 20 of GLP-1 showed minimal impact on Ala. The Ala substitution at positions 10 and 11 of GIP has almost no negative impact on insulin promoting activity, while mutations at positions 12 and 14 have a greater negative impact. Ile12 plays a crucial role in the activation of GIPR 15. Tyr10 and Ile12 are also used in tilposide. In GLP-1 (7-37) NH2, there was a slight increase in affinity and activity when Tyr16 was used instead of Val16. Therefore, tigapamide may slightly reduce GLP1 activity but enhance GIP activity.
Aib13
GLP-1, GIP, GCG (glucagon), and GLP-2 have high homology, therefore it should be considered that the designed GLP-1/GIP dual agonists should avoid the activation of GCGR and GLP-2R. In the experiment, replacing position 13 with Aib made the agonist inactive towards GCGR and slightly altered the activities of GLP-1 and GIP. Replacing GLP-1 (7-37) Tyr19 with Ala significantly reduced affinity and activity.
In GIP, the possibility of replacing Ala13 with Aib has little impact on activity. The Aib13 of texaparide seems to reduce its GLP-1 activity without affecting its GIP activity and disrupt its GCG activity, but this hypothesis lacks supporting evidence. However, Aib13 is unnecessary as other alternatives can play the same role. Furthermore, considering the Gln13 in exenatide, GLP-1 may interact with GLP-1R through hydrogen bonding. Using Tyr, Gln, Thr, Ser, and Lys can also be worth trying in this position.
Ala18:
The mutation of GIP His18 to Ala enhances its insulin promoting effect. Replacing Arg18 in GCG with alanine simultaneously increased the activity of GLP-1 and GIP. The use of Ala18 from GLP-1 may increase the probability of GIP activity.
Lys20-X:
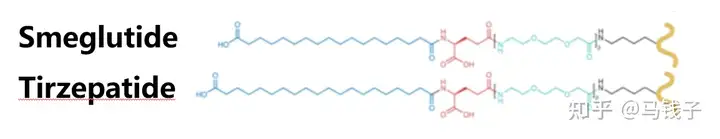
Replacing GLP-1 Lys26 with Ala will result in a slight decrease in affinity and activity. In semaglutide, fatty acid chains are connected at this site, enhancing affinity and activity. GLP-1 Lys26 may form polar interactions with receptors, which may be enhanced by Arg20 in exenatide, but does not result in steric hindrance. GIP is similar, as GIP-Gln20 appears to form hydrogen bonds with GIPR-Asn120 and is unobstructed in the extracellular matrix. The fatty acid chain connected to Lys20 in Tilposide is similar in structure to Smeglutide, except for two additional carbon chains in the middle, with the junction consistent with the end. Research results show that it can improve GLP-1 activity, while GIP activity is not affected, and more importantly, it prolongs Tilposide absorption and bolism.
Ala21:
The mutation of GLP-1 Glu27 to Ala does not change affinity, but slightly increases activity. In the experiment, Asp21 of GIP was replaced by Ala; GLP-1 activity showed minimal changes, but a slight decrease was observed in GIP activity. Therefore, Glu27 of GLP-1 is not a key point in the interaction with GLP-1R. Without GIP (31-42) tail, choosing Ala21 may increase GLP-1 activity, although it may slightly decrease GIP activity.
Val23, Ile27:
GLP-1 (7-37) Phe28, Trp31, and Leu32 are symmetric with GIP's 22, 25, and 26 bits. GLP-1, GIP, and exenatide seem to have the same pattern here: Phe22 interacts specifically with the receptor in the center, and adjacent hydrophobic residues at positions 23, 25, and 26 are used to form hydrophobic regions. The residue at position 24 further away can interact with receptor polarity and may be beneficial for locating hydrophobic regions. GLP-1 Ile29 and Val33 have properties similar to GIP Val23 and Leu27. The mutation of GLP-1 Phe28 to Ala resulted in almost all of its affinity loss, and the Ile29 mutation significantly reduced affinity, while the Trp31, Leu32, and Val33 mutations had almost no effect, indicating that they may only be secondary. The use of Val23 and Ile27 in Tilposide may have little effect on GLP-1 or GIP activity. Because they are the same residue and no specific interactions have been detected at these sites.
Gln24:
Replacing GLP-1 Ala30 with Gln reduced affinity but increased activity. GIP Asn24 is similar to Gln; Therefore, the use of Gln24 in Tilposide can enhance GLP-1 activity and support GIP activity.
Ile27:
From positions 33-35 of GLP-1, with each substitution of Ala, the IC50 increases approximately fivefold, but the EC50 slightly decreases. Although these substitutions reduce affinity, ligands are used to "correct" the structure. Connect positions 30 and 34 with the lactam bridge to enhance both affinity and activity. Structural 'correction' may mean the formation of a more stable α- Spiral. Replacing GLP-1 Arg36 with Ala significantly hindered affinity and activity. If Arg36 of GLP-1 is removed, both affinity and activity will decrease by 10 times, indicating that it is crucial for interactions. In addition, Gly35, Arg36, and Gly37 may contribute to the selectivity of GLP-1.
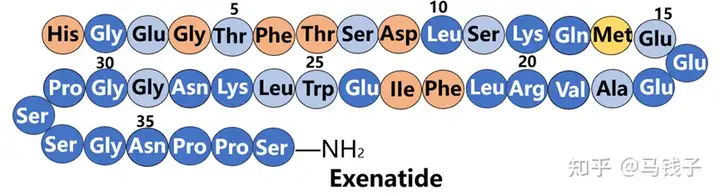
Compared to GLP-1, exenatide has 9 additional residues at the C-terminus, which form the so-called "Trp cage" structure, folding and protecting Trp from exposure. As the C-terminus is truncated, exenatide is exposed to NEP 24.11 degradation. In addition, connecting the C-terminal of GLP-1 to exenatide (31-39) can prevent DPP-4 degradation. As the C-terminal decreases, the proinsulin activity of exenatide gradually decreases. Interestingly, when it was reduced to exenatide (1-28), its proinsulin activity almost regained all integrity, but when it reached exenatide (1-26), its proinsulin activity suddenly disappeared. Obviously, Lys27 and Asn28 of exenatide play an important role in binding to receptors. Most of the previous experiments were a comparison between exenatide (1-30) and GLP-1, exploring the function of the additional C-terminal residue (31-39) of exenatide. However, in the above experiment, a comparison should be made between exenatide (1-28) and GLP-1, as exenatide (29-39) is indeed an additional fragment. At the same time, the secondary structure of the peptide and therefore the conformation for receptor recognition are more important.
Compared with the first generation GLP-1R agonist, Tilposide, as a GLP-1/GIP dual receptor agonist, has three key improvements: firstly, many residues in the peptide skeleton are altered to obtain GIPR activation activity; Secondly, the C-terminal sequence of exenatide extends the C-terminal; Thirdly, a fatty acid side chain is conjugated, similar to a semaglutide prolonging half-life. By studying the different residues used at each position, the activity of dual agonists can be customized. Initially, the main focus was on receptor activation activity, but recently, the downstream effects of GLP-1R and GIPR have increasingly shown that receptor activation not only promotes cAMP and stimulates insulin release, but also other factors( β- Inhibiting protein recruitment, residence time, etc. can also affect receptor internalization, further triggering long-term effects in the body.
Existing research has shown that in comparison with the hypoglycemic effect of smeagllutide, telopoptide reduces subjects' glycated hemoglobin by an average of 2.0-2.3%, while smeagllutide reduces subjects' glycated hemoglobin by an average of 1.9%; In terms of weight loss, telopoptide resulted in an average weight loss of 17 to 25 pounds in participants, while smeglutide was 13 pounds. Tilposide exhibits superior levels of hypoglycemic and lipid-lowering properties compared to smeglutide. The safety of tilposide is similar to that of other enterotropin drugs. The gastrointestinal reactions of tilposide, such as nausea, diarrhea, and constipation, were significantly improved compared to placebo, but most of them were mild to moderate. Other adverse reactions, especially hypoglycemia, have not been found to have significant differences between the two groups.
With the widespread use of GLP-1 analogues in the treatment of T2DM and weight loss populations, intestinal stimulating insulin (GIP) appears mysterious and has limited clinical application. However, scientists have never stopped exploring, and the outstanding clinical effects of Tilposide's successful launch are a reward for these efforts.
reprint :https://zhuanlan.zhihu.com/p/641902921?utm_id=0
1.M. Yu, et al., Battle of GLP-1 delivery technologies, Adv. Drug Deliv. Rev. (2018)
2.Lijing Wang, Designing a Dual GLP-1R/GIPR Agonist from Tirzepatide: Comparing Residues Between Tirzepatide, GLP-1, and GIP Drug Design, Drug Design, Development and Therapy 2022:16 1547–1559